|
|
Гіпогонадизм
Общая информацияГіпогонадизм – поліетіологічне захворювання, в основі якого лежить
відсутність або зниження здатності статевих залоз продукувати адекватну
кількість статевих гормонів.
Відрізняють первинний (гіпергонадотропний) і вторинний (гіпогонадотропний)
гіпогонадизм.
В основі гіпергонадотропного (первинного) гіпогонадизму лежить ураження статевих
залоз (яєчок, яєчників), у результаті чого первинно порушується їх здатність
продукувати статеві гормони.
При гіпогонадотропному гіпогонадизмі має місце відсутність чи зниження здатності
гіпоталамусу секретувати гонадоліберин або гіпофізу – секретувати ЛГ, ФСГ, у
результаті чого вторинно знижується здатність периферичних статевих залоз
продукувати статеві гормони.
Етіологія
Причиною гіпогонадизму є вроджені чи набуті аномалії розвитку яєчок
і яєчників (первинна форма), або ЦНС та гіпоталамо-гіпофізарних структур (вторинна
форма).
Вроджені аномалії розвитку гонад, ЦНС та гіпоталамо-гіпофізарних структур у
більшості випадків пов’язані з хромосомними дефектами та генними мутаціями.
Набуті форми гіпергонадотропного гіпогонадизму виникають у результаті
аутоімунних та інфекційних захворювань яєчок та яєчників, їх травм, хірургічних
втручань, променевої або хіміотерапії, прийому протипухлинних препаратів,
наркотиків, токсичних речовин тощо.
Набуті форми гіпогонадотропного гіпогонадизму виникають у результаті розвитку
пухлин гіпоталамо-гіпофізарної області та ЦНС, інфекційних процесів (туберкульоз,
менінгоенцефаліт, грип, та ін.), хірургічних втручань з приводу пухлин гіпофізу,
черепно-мозкових травм, опромінення гіпоталамо-гіпофізарної ділянки тощо.
Вторинний гіпогонадизм розвивається у хворих на велику таласемію та гемохроматоз
(багатократні переливання крові викликають відкладання заліза в гіпофізі та
гіпоталамусі).
Певну роль у виникненні обох форм гіпогонадизму відіграють такі чинники:
патологія вагітності та пологів у матері, гіпоксія плоду, важкі соматичні та
ендокринні захворювання у дитини.
Патогенез
При гіпергонадотропному гіпогонадизмі у первинно уражених гонадах
порушується синтез і секреція статевих стероїдів, рівень їх у циркулюючій крові
знижується і, відповідно до механізму зворотного зв’язку, відбувається
гіперстимуляція аденогіпофізу, що призводить до підвищення рівня гонадотропінів.
В основі гіпогонадотропного гіпогонадизму лежить порушення прямого
стимуляційного (позитивного) зв’язку в системі гіпоталамус-гіпофіз-гонади або в
системі гіпофіз-гонади. Порушення секреції гонадоліберину в гіпоталамусі
призводить до зниження продукції ЛГ та ФСГ у гіпофізі, в результаті чого
секреція статевих стероїдів у периферичних статевих залозах також знижується.
При ураженні тільки гонадотропних клітин гіпофізу знижується продукція ЛГ, ФСГ
та статевих гормонів; функція гіпоталамусу при цьому не страждає.
Ізольований дефіцит гонадотропних гормонів майже завжди обумовлений хронічною
недостатністю гонадоліберину й лише в деяких випадках – недостатністю
гонадотропних клітин гіпофізу.
Первинне ураження гонадотропних клітин гіпофізу часто поєднується з
недостатністю інших тропних гормонів (найчастіше – з недостатністю СТГ та ТТГ). Клиническая картинаОбумовлена зниженням рівнів статевих гормонів.
У дітей дефіцит статевих гормонів призводить до затримки статевого дозрівання (у
хлопців – відсутність ознак статевого дозрівання після 13,5-14 років, у дівчат –
відсутність телархе та адренархе у 13-14 років, менархе – у 15-16 років). Часто
статевий розвиток починається, але не завершується.
У хлопчиків спостерігається різний ступінь затримки розвитку зовнішніх геніталій
вже в допубертатному періоді. Яєчка зменшені у розмірах, за консистенцією –
в’ялі або ущільнені. Часто спостерігається крипторхізм. Статевий член менший за
розмірами таких, що характерні для вікової норми, у деяких випадках
спостерігається мікропеніс.
Якщо дефіцит тестостерону або резистентність тканин-мішеней до андрогенів
виникає у перші 14-16 тижнів ембріогенезу, то у новонароджених хлопчиків
формуються зовнішні статеві органи змішаного типу (впритул до розвитку
нормальних жіночих зовнішніх статевих органів та скороченої вагіни – чоловічий
несправжній гермафродитизм).
У пубертатному періоді не спостерігається збільшення об’єму яєчок (об’єм яєчок
не перевищує 3-4 мл); не розвиваються, або слабко розвиваються, вторинні статеві
ознаки: відсутні мутація голосу і оволосіння на обличчі, ювенільне акне, ерекції
та полюції.
У постпубертатному періоді зовнішні та внутрішні геніталії залишаються
недорозвиненими; оволосіння – рідке, характерним є його розподіл за жіночим
типом; погано розвинені скелетні м’язи; з’являються еректильні дисфункції,
олігозооспермія або азооспермія.
У дівчат спостерігається відсутність розвитку молочних залоз, перерозподілу жиру
за жіночим типом, первинна або вторинна аменорея. Зовнішні статеві органи
інфантильні, яєчники та матка зменшені у розмірах (іноді – різко гіпоплазовані
або рудиментарні), у яєчниках знаходять переважно примордіальні фолікули.
Лобкове та акселярне оволосіння у хворих обох статей може бути відсутнім, дуже
слабко розвиненим, або не перевищувати 3 стадії за Танером (якщо не порушена
секреція наднирникових андрогенів, які обумовлюють адренархе).
У більшості підлітків, які страждають на гіпогонадизм, швидкість росту у
препубертатний період буває нормальною, або ж її незначне зниження
спостерігається у пубертатному періоді в результаті відсутності ростового
стрибка. Здебільшого кінцевий ріст хворих на гіпогонадизм не страждає і навіть
перевищує генетично детермінований, оскільки зони росту довго залишаються
відкритими, а спроможність до лінійного росту зберігається до 18-20 років та
більше. Кістковий вік у таких пацієнтів спонтанно досягає межі 13-14 років у
хлопчиків, 11,5-12,5 років – у дівчат, і тільки після цього відмічається
затримка кісткового дозрівання. У постпубертатний період така затримка може
виявитися дуже суттєвою.
Виключення становлять синдроми Шерешевського-Тернера, Нунан, тернероїдна форма
змішаної дизгенезії гонад, множинний дефіцит тропних гормонів гіпофізу та деякі
інші синдроми, що супроводжуються гіпогонадизмом. У таких дітей значна затримка
росту й кісткового віку спостерігається вже у допубертатному періоді; без
додаткового лікування хворі залишаються низькорослими.
У хворих на гіпогонадизм вже у пубертатному віці починають формуватися
євнухоїдні, геноїдні або інфантильні пропорції тіла та остеопороз.
Характеристика окремих форм гіпогонадотропного гіпогонадизму
Синдром Кальмана
Синдром Кальмана є найчастішою причиною гіпогонадотропного гіпогонадизму у
хлопчиків. Його частота коливається у межах 1:5000-10000 чоловіків у популяції.
У дівчат синдром Кальмана зустрічається значно рідше.
Синдром Кальмана обумовлений вадами розвитку переднього мозку, зокрема –
порушенням міграції гонадоліберинсекретуючих нейронів у ольфакторні цибулини й у
подальшому – в гіпоталамус. Ці анатомічні аномалії призводять до дефіциту
гонадоліберину або до порушення імпульсного ритму його секреції.
Синдром Кальмана – спадкове захворювання з аутосомно-домінантним,
аутосомно-рецесивним або Х-зчепленим характером спадковості, виявлені дефекти
гену KALIG-1 (KAL1) на Хр22.3 хромосомі, дефекти гену FGFR1 (KAL2) на короткому
плечі 8 хромосоми.
Характерною особливістю синдрому є поєднання гіпогонадизму з порушенням нюху
(аносмія або гіпоосмія).
Крім того, при синдромі Кальмана можуть зустрічатися й інші аномалії
розвитку. Вони обумовлені, в першу чергу, дефектами формування ЦНС: спастичні
параплегії, туговухість, горизонтальний ністагм, порушення кольорового зору,
незарощення піднебіння та верхньої губи, затримка розумового розвитку. Можлива
наявність симптомів, які пов’язані з вадами розвитку сечостатевої системи:
агенезія нирок, підковоподібна нирка, у хлопчиків – крипторхізм та мікропеніс.
Клінічні прояви можуть бути досить варіабельними. Навіть хворі діти з
однієї сім’ї можуть мати різні клінічні прояви синдрому: від легкої гіпоосмії,
яка виявляється тільки спеціальними методами, і нормального статевого розвитку,
до вираженої аносмії та важкого гіпогонадизму, що супроводжується іншими вадами
розвитку.
Хворі на синдром Кальмана високорослі, мають євнухоїдні пропорції тіла.
Відставання кісткового віку від паспортного починається у 13-14 років. Зони
росту залишаються відкритими до 20 років і більше.
Гормональні ознаки: зниження рівнів ЛГ, ФСГ, тестостерону (у хлопчиків),
естрадіолу (у дівчат); при проведенні стимуляційного тесту з гонадореліном –
рівень ЛГ та ФСГ не перевищує допубертатних значень ( 10 Од/л). Стимуляційний
тест з ХГ – позитивний.
Біля 10% підлітків, хворих на синдром Кальмана, мають парціальну форму
гіпогонадотропного гіпогонадизму – за первинною діагностикою в 14-15 років
підвищення ЛГ на стимуляцію гонадореліном буде досягати пубертатних значень,
однак у подальшому зрілий рівень гонадотропних та статевих гормонів не
досягається.
Фертильний євнухоїдний синдром (синдром фертильного євнуха, синдром
Паскуаліні, ізольований дефіцит ЛГ)
Дуже рідкісне захворювання. Вважається, що причиною синдрому є ізольований
дефіцит ЛГ або частковий дефіцит гонадоліберину. У результаті в яєчках
порушується проліферація та диференціювання клітин Лейдіга. Поодинокі клітини
Лейдіга секретують тестостерон у кількостях, що є достатніми для підтримки
сперматогенезу, але не достатніми для повної андрогенізації.
Клінічні прояви: затримка статевого дозрівання, євнухоїдна будова тіла,
високорослість. Відставання кісткового віку від паспортного починається у 13-14
років. Зони росту залишаються відкритими до 18-20 років.
Яєчка – нормальних розмірів, при біопсії диференційовані клітини Лейдіга не
виявляються, або їх дуже мало, але сперматогенез не порушений, тому хворі не
безплідні.
Гормональні ознаки: базальний та стимульований гонадореліном рівень ЛГ
знижений або знаходиться на нижній межі норми, рівень ФСГ нормальний, рівень
тестостерону – знижений. Стимуляційний тест з ХГ – позитивний.
Вроджені множинні поєднані дефекти розвитку
Синдром Прадера-Віллі
Захворювання має аутосомно-домінантний тип успадкування і обумовлене
делецією ділянки проксимального відділу довгого плеча 15 хромосоми батьківського
походження або подвоєнням 15 хромосоми, яке виникає у результаті дефекту
розходження материнської хромосоми. Встановлено, що пацієнти з делецією
батьківської хромосоми мають типові фенотипічні ознаки синдрому, а пацієнти з
подвоєнням материнської хромосоми мають стерту форму захворювання. Частота
синдрому у популяції становить 1:25000. 70-80% пацієнтів мають делецію 15
хромосоми.
Механізм розвитку гіпогонадизму до кінця не з’ясований. Припускається патологія
гіпоталамусу.
Клінічні ознаки. Новонароджені діти мають виражену м’язову гіпотонію,
низькі показники ваги та зросту. Типовими для синдрому є маленькі розміри кистей
та стоп з укороченими пальцями. Форма обличчя характеризується звуженим
фронтальним діаметром, очі – близько розташовані, міміка – слабко виражена. У
процесі росту дитини м’язова гіпотонія стає менш вираженою, на перший план
виступає булімія, яка супроводжується вираженим ожирінням. У підлітковому віці
надлишок калорій призводить до розвитку цукрового діабету; значний надлишок ваги
супроводжується серцевою недостатністю, яка часто є причиною ранньої смерті
пацієнтів. Для синдрому Прадера-Віллі характерна низькорослість, у більшості
випадків спостерігається розумова відсталість.
Порушення статевого розвитку у хлопчиків зі синдромом Прадера-Віллі виявляють
досить рано у вигляді крипторхізму та мікропенісу. Клінічні симптоми
гіпогонадизму у дітей обох статей проявляються у пубертатному віці.
Гормональні ознаки: зниження рівнів ЛГ, ФСГ, тестостерону (у хлопчиків),
естрадіолу (у дівчат); при стимуляційному тесті з гонадореліном рівень ЛГ та ФСГ
не перевищує допубертатних значень (< 10 Од/л). Стимуляційний тест з ХГ –
позитивний.
У окремих дітей може спостерігатися ураження гонад. У таких випадках рівні
гонадотропінів можуть підвищуватись.
Синдром Лоуренса-Муна-Барде-Бідля
Захворювання має аутосомно-рецесивний тип успадкування. З найбільшою
частотою зустрічається у арабській популяції Кувейту (1:13000).
Генетична природа синдрому до теперішнього часу не встановлена.
Механізм розвитку гіпогонадизму до кінця не з’ясований. Припускається патологія
гіпоталамусу.
Клінічні ознаки. Найхарактернішими ознаками, що зустрічаються практично у
всіх пацієнтів, є ожиріння, пігментний ретиніт, тотальний або парціальний
гіпогонадизм.
Ураження органів зору, окрім пігментного ретиніту, характеризуються атиповою
дрібнокрапковою ретинопатією, макулярною дегенерацією, офтальмоплегією,
мікрофтальмією, міопією.
Полі- та синдактилія спостерігається у 75% хворих.
У 70% дітей зустрічаються вроджені аномалії нирок, серед яких найбільш поширеною
є кістозна дисплазія.
Розумова відсталість спостерігається у 50% пацієнтів та проявляється у діапазоні
від легкої дебільності до ідіотії.
У частини хворих розвивається цукровий або нецукровий діабет.
Порушення статевого розвитку у переважної більшості хлопчиків із синдромом
Лоуренса-Муна-Барде-Бідля виявляються досить рано у вигляді різкого недорозвитку
зовнішніх статевих органів. Клінічні симптоми гіпогонадизму у дітей обох статей
проявляються у пубертатному віці.
Гормональні ознаки: зниження рівнів ЛГ, ФСГ, тестостерону (у хлопчиків),
естрадіолу (у дівчат); при стимуляційному тесті з гонадореліном рівень ЛГ та ФСГ
не перевищує допубертатних значень (< 10 Од/л). Стимуляційний тест з ХГ –
позитивний.
Характеристика окремих форм гіпергонадотропного гіпогонадизму
Синдром Клайнфельтера
Синдром Клайнфельтера є найчастішою причиною розвитку гіпергонадотропного
гіпогонадизму у хлопчиків. Частота синдрому у популяції становить 1:300-1:1000
хлопчиків.
Захворювання обумовлено хромосомною патологією – полісомією за Х-хромосомою. Для
класичного варіанту синдрому Клайнфельтера є характерним каріотип 47ХХY. У 10%
хворих спостерігається мозаїцизм 46ХY/47ХХY, 47ХХY/46ХХ, 47ХХY/45ХО. Значно
рідше зустрічаються каріотипи 48ХХХY, 49ХХХХY, 47ХХY/46ХХ/45ХО. Наявність
додаткової Х-хромосоми у чоловічому каріотипі може пояснюватися нерозходженням
Х-хромосоми у період першого або другого мейотичного ділення або порушенням
мітотичного розходження хромосом у період розвитку зиготи (мозаїчні варіанти).
Клінічні ознаки. До пубертатного віку у хлопчиків можуть виявлятися
крипторхізм (частіше – двобічний), маленькі розміри яєчок та статевого члена. У
50% хлопчиків відмічається помірна затримка розумового розвитку, яка
супроводжується порушенням поведінки, важкістю контактів з однолітками.
У пубертатному віці вторинне оволосіння з’являється у звичайні строки,
відмічається також збільшення статевого члена. Однак об’єм тестикул збільшується
незначно, не перевищуючи, як правило, 8 мл. Яєчка мають ущільнену консистенцію.
Фенотип у хворих – чоловічий, характерні високорослість, непропорційно довгі
ноги, євнухоїдна будова тіла, гінекомастія.
Відставання кісткового віку від паспортного починається у 13-14 років. Зони
росту залишаються відкритими до 18-20 років.
Вторинне оволосіння незначне, за жіночим типом. У постпубертатному періоді яєчка
залишаються маленькими (довга вісь – менше 2 см), ущільненими. При біопсії яєчка
знаходять гіаліноз сім’яних канальців, зменшення числа або відсутність клітин
Сертолі; сперматогенез – відсутній.
За наявності мозаїцизму спостерігаються менш важкі порушення. У чверті хворих з
мозаїцизмом яєчка мають нормальні розміри. Сперматогенез відсутній тільки у
половини хворих.
Окрім симптомів порушення статевого розвитку, у хворих на синдром Клайнфельтера
може виявлятися ряд вроджених аномалій кісткової тканини: клінодактилія,
деформація грудини, вальгусні деформації ліктьових та колінних суглобів,
гіпертелоризм, мікрогнатія, готичне піднебіння та ін. Нерідко захворювання
супроводжується вродженими вадами серцево-судинної системи. Часто
спостерігаються захворювання щитоподібної залози, цукровий діабет, хронічні
обструктивні захворювання легенів.
У чоловіків з синдромом Клайнфельтера ризик раку молочної залози у 20 разів
вищий, ніж у чоловіків з нормальним каріотипом.
Гормональні ознаки. У допубертатному віці показники ЛГ, ФСГ та
тестостерону зазвичай відповідають нормі. На початку пубертату рівень ФСГ
підвищується і у
14-15 років вже значно перевищує норму. Рівень тестостерону на початку пубертату
зазвичай підвищується, але не досягає нормативних показників. Рівень ЛГ у
пубертатний період відповідає нормі, але в подальшому концентрація ЛГ
підвищується. У пубертатному періоді спостерігається відносна гіперестрогенія.
Реакція ЛГ та ФСГ на введення гонадореліну носить гіперергічний характер уже на
ранніх стадіях пубертату.
Для підтвердження або уточнення діагнозу визначають статевий хроматин (у хворих
на синдром Клайнфельтера він позитивний), проводять каріотипування.
При мозаїчному варіанті синдрому хромосомні аномалії можуть локалізуватися
тільки у клітинах яєчок, тому діагноз підтверджують за допомогою біопсії яєчок.
Синдром Шерешевського-Тернера
Синдром Шерешевського-Тернера (СШТ) є найчастішою причиною розвитку
гіпергонадотропного гіпогонадизму у дівчат.
Частота синдрому у популяції становить 1:2000-1:5000 дівчат.
Захворювання обумовлено хромосомною патологією – за класичним варіантом синдрому
– це моносомія Х-хромосоми. Для класичного варіанту синдрому
Шерешевського-Тернера є характерним каріотип 45ХО (50-60% хворих). У 20%
випадків спостерігається мозаїцизм 45ХО/46ХХ або 45ХО/46ХY. В інших випадках
зустрічаються структурні аномалії Х-хромосоми: делеції короткого або довгого
плеча Х-хромосоми, кільцева Х-хромосома, ізохромосома Х. Іноді спостерігаються
транслокації між Х-хромосомою та аутосомами. Кількісні та структурні аномалії
Х-хромосоми можуть бути обумовлені порушенням мейотичного розходження хромосом.
Клінічні ознаки. Найтиповішими для СШТ є низькорослість, статевий
інфантилізм, множинні вроджені аномалії розвитку. Слід пам’ятати, що
зустрічаються варіанти синдрому без виражених множинних аномалій розвитку.
У період новонародженості характерна наявність лімфатичних набряків стоп,
кистей, верхньої частини тулуба та шиї.
У старшому віці звертає на себе увагу характерний фенотип, який формується у
результаті поєднання низькорослості (починає формуватися з 3-4 років) та
вроджених аномалій розвитку. Для СШТ характерними є: бочкоподібна або плоска
грудна клітка з широко розташованими сосками, коротка шия з характерними «крилоподібними»
складками, низько розташовані деформовані вушні раковини, низька межа росту
волосся на потилиці, «антимонголоїдний» розріз очей, епікант, птоз вій, «готичне
піднебіння», мікрогнатія, множинні пігментні невуси, укорочення IV і
V п’ясних або преплюсневих кісток, гіпоплазія нігтів, вальгусна деформація
ліктьових та колінних суглобів, різноманітні порушення розвитку сечовидільної та
серцево-судинної систем, сенсорні порушення слуху.
Порушення статевого розвитку проявляються повною відсутністю розвитку молочних
залоз, мізерним аксилярним та лобковим оволосінням, первинною аменореєю.
Зовнішні статеві органи сформовані за жіночим типом, недорозвинені. Матка –
різко гіпоплазована, маточні труби – тонкі або рудиментарні, яєчники –
дизгенетичні (можуть мати різну кількість примордіальних фолікулів) або
представлені тяжеподібними формуваннями, які не мають ооцитів 1 порядку та
фолікулів.
Для СШТ характерним є негативний або дуже низький статевий хроматин (3-5%).
У хворих на СШТ з мозаїцизмом 45ХО/46ХY, а також у пацієнтів без Y-хромосоми,
але з наявністю гена SRY (фактор, що кодує розвиток яєчка; виявляється за
допомогою молекулярно-генетичного дослідження), у зачатках статевих залоз може
знаходитись гормонально активна тканина яєчок, у зв’язку з чим зростає ризик
розвитку гонадобластоми.
Гормональні ознаки. СШТ характеризується високими рівнями ЛГ і ФСГ у
крові та низькими рівнями естрогенів (особливо – естрадіолу) у крові та сечі.
Підвищення рівнів гонадотропних гормонів, особливо ФСГ, спостерігається вже у
перші тижні життя та зберігається протягом 1,5-3 років. З 3-4 до 5-7 років
відмічається зниження рівнів гонадотропінів до вікової норми, що пов’язано з
активацією універсального центрального механізму, який пригнічує імпульсну
секрецію гонадоліберину, незалежно від рівнів статевих стероїдів. З 5-6 років
концентрація ЛГ, а особливо – ФСГ, знову починає збільшуватися. Значне
підвищення їх рівнів, яке може майже у 10 та більше разів перевищувати норму,
спостерігається на початку пубертатного віку.
Реакція ЛГ та ФСГ на введення гонадореліну носить гіперергічний характер уже на
ранніх стадіях пубертату.
Синдром Нунан
Синдром Нунан спостерігається в осіб обох статей; має аутосомно-домінантний
тип спадковості або зустрічається спорадично. Вважається, що захворювання
обумовлено експресією патологічного гена, який локалізується на довгому плечі 12
хромосоми.
Клінічні ознаки. Всі хворі на синдром Нунан мають виражені симптоми
дисембріогенезу. Зовнішні клінічні ознаки (низькорослість, коротка шия з «крилоподібними»
складками, лімфатичні набряки кистей та стоп, вальгусна деформація суглобів, «трикутне
обличчя», «антимонголоїдний» розріз очей, епікант, птоз вій, «готичне піднебіння»,
деформації грудної клітки тощо) схожі з синдромом Шерешевського-Тернера. Типовим
для синдрому Нунан є наявність у хворих кардіоваскулярних вад розвитку: стеноз
легеневої артерії (80% хворих), гіпертрофія міжшлуночкової перетинки та ін.
Характерною ознакою синдрому Нунан, що відрізняє його від СШТ, є наявність
нормального каріотипу у хворих (в чоловіків – 46ХY, у жінок – 46ХХ). Як наслідок
– статевий хроматин відповідає генетичній статі.
У хлопчиків спостерігається статевий інфантилізм, часто має місце
крипторхізм, гіпоплазія яєчок, зустрічається мікропеніс. У пубертатному віці
формуються біохімічні ознаки гіпергонадотропного гіпогонадизму. У більшості
дорослих чоловіків порушена фертильність.
У дівчат в пубертатному віці спостерігається затримка статевого дозрівання. Але
функція яєчників, у переважній більшості випадків, не порушена, в результаті
чого статева і репродуктивна функція у дорослих жінок страждає не суттєво.
Гормональні ознаки. У хлопчиків спостерігається високий рівень ЛГ, ФСГ та
низький рівень тестостерону. Тест із введенням ХГ – негативний. Реакція ЛГ та
ФСГ на введення гонадореліну носить гіперергічний характер уже на ранніх стадіях
пубертату.
Чиста дизгенезія гонад
Синдром «чистої» дизгенезії гонад зустрічається у осіб з нормальним
чоловічим (46ХY) або жіночим (46ХХ) каріотипом і характеризується наявністю
жіночого фенотипу у всіх випадках (незалежно від генетичної статі), різко
вираженим статевим інфантилізмом, відсутністю множинних соматичних аномалій
розвитку (останній факт пояснює назву «чиста» дизгенезія).
В основі синдрому лежать крапкові мутації генів на Х-хромосомі (за наявності
каріотипу 46ХХ), або мутаціями гена SRY на Y-хромосомі (за наявності каріотипу
46ХY).
Частота синдрому становить 1:25000-1:35000 осіб.
Клінічні ознаки. «Чиста» дизгенезія гонад зазвичай діагностується у
пубертатному та постпубертатному віці, коли хворі звертаються з приводу
відсутності або різкої затримки статевого дозрівання, первинної аменореї.
Всі хворі мають жіночий фенотип, нормальний зріст у препубертатному та на
початку пубертатного віку. З 13-14 років починають формуватися високорослість,
євнухоїдні пропорції тіла.
Відставання кісткового віку від паспортного становить, у середньому, 3-5 років і
є результатом вираженої гіпоестрогенії.
Кінцевий зріст у більшості хворих перевищує 170 см; іноді – залишається середнім.
Звертає на себе увагу повна відсутність або різке недорозвинення молочних залоз,
мізерне аксилярне та лобкове оволосіння.
Зовнішні та внутрішні статеві органи недорозвинені. Матка та маточні труби –
різко гіпоплазовані або рудиментарні. У всіх хворих спостерігається первинна
аменорея.
Яєчники представлені тяжеподібними формуваннями, які не містять ооцитів і
фолікулів (або містять поодинокі примордіальні фолікули).
У хворих з каріотипом 46ХY у дизгенетичних гонадах можуть бути виявлені зародки
сім’яних канальців та клітин Лейдіга. Такі пацієнти мають дуже великий ризик
розвитку дизгерміноми або гонадобластоми.
Гормональні ознаки. Характерні високі рівні ЛГ та ФСГ у крові й низькі
рівні естрогенів (особливо естрадіолу) у крові та сечі.
Реакція ЛГ й ФСГ на введення гонадореліну носить гіперергічний характер уже на
ранніх стадіях пубертату.
Змішана дизгенезія гонад (змішана дизгенезія яєчок, змішаний гонадальний
дизгенез) (див. розділ «Порушення статевого диференціювання»).
Одна з найпоширеніших форм несправжнього чоловічого гермафродитизму (складає
25-30% від усіх форм несправжнього чоловічого гермафродитизму). Характеризується
наявністю дизгенетичного яєчка, яке розташовується у черевній порожнині,
паховому каналі або у рудиментарній мошонці; стрека – недиференційованого
сполучнотканинного тяжу, що розташовується у черевній порожнині з іншого боку (у
деяких випадках стрек може бути відсутнім) та матки з однією або обома маточними
трубами.
У більшості хворих на змішану дизгенезію гонад (ЗДГ) виявляється мозаїчний
каріотип 45ХО/46ХY, зустрічається також нормальний чоловічий каріотип – 46ХY,
рідше – інші форми мозаїцизму.
Клінічні ознаки. Будова зовнішніх і внутрішніх геніталій у хворих на ЗГД
залежить від клону клітин, який переважає у каріотипі та від ступеня ураженості
Y-хромосоми.
Для пацієнтів, у яких превалює клон ХО, Y-хромосома суттєво пошкоджена, а яєчко
не функціонувало у внутрішньоутробному періоді, характерна більш жіноча будова
зовнішніх геніталій (іноді спостерігається тільки гіпертрофія клітора).
Внутрішні геніталії представлені гіпоплазованими маткою та трубами, присутня
вагіна або верхня її частина. Яєчко розташоване у черевній порожнині, має ознаки
дизгенезії.
Такі пацієнти низькорослі, за фенотипом схожі на хворих, які страждають на
синдром Шерешевського-Тернера.
Для пацієнтів, у яких превалює клон ХY, Y-хромосома не так суттєво пошкоджена, а
яєчко функціонувало у внутрішньоутробному періоді, характерна змішана будова
зовнішніх геніталій. Спостерігається урогенітальний синус, статевий член нагадує
гіпертрофований клітор, мошонка розщеплена, у зморшках. Внутрішні геніталії
представлені різко гіпоплазованою маткою та, здебільшого, єдиною трубою;
найчастіше присутня верхня частина вагіни. Дизгенетичне яєчко розташоване у
паховому каналі або у рудиментарній мошонці. Збоку яєчка розвивається придаток
яєчка, сім’явиносна протока.
У деяких випадках фенотип і зовнішні статеві органи бувають ближчими до
чоловічих.
Такі хворі можуть мати нормальний або високий зріст.
У всіх пацієнтів єдине яєчко має ознаки дизгенезії різного ступеня,
спостерігається порушення його сперматогенної функції, але у частини хворих
можливість яєчка продукувати тестостерон зберігається («функціонуюче яєчко»).
Унаслідок здатності яєчка продукувати тестостерон у пубертатному періоді часто
спостерігається статеве дозрівання з вираженою андрогенізацією. У переважній
більшості випадків статеве дозрівання буває неповним.
Слід пам’ятати, що у хворих на СДГ існує дуже великий ризик розвитку
злоякісних пухлин із дизгенетичного яєчка, а поява симптомів андрогенізації може
бути першою ознакою розвитку пухлини.
Хворі на СГД мають негативний, або дуже низький, статевий хроматин.
Гормональні ознаки. Залежать від функціональної можливості яєчка. Рівень
ЛГ та ФСГ може бути незначно підвищеним або високим. Вміст тестостерону
коливається у межах від низького до нормального (якщо функціональна активність
яєчка збережена).
При позитивному тесті з ХГ можливий спонтанний пубертат.
Синдром тестикулярної фемінізації (див. розділ «Порушення статевого
диференціювання»).
Синдром тестикулярної фемінізації (СТФ) є однією з найчастіших причин
несправжнього чоловічого гермафродитизму. Частота синдрому становить 1:50000
осіб. Захворювання має Х-зчеплений характер спадковості. Пацієнти мають
негативний статевий хроматин, каріотип 46ХY і жіночий фенотип.
Патогенез СТФ обумовлений дефектом гену рецептору андрогенів (локалізується на
короткому плечі Х-хромосоми) і характеризується нечутливістю тканин до
андрогенів.
Клінічні ознаки. Існує два основних клінічних варіанти СТФ – повна форма
(синдром Моріса), за умов якої зовнішні геніталії мають нормальну жіночу будову,
і неповна, при якій зовнішні геніталії мають різний ступінь маскулінізації.
Похідні мюллерових протоків (матка, фалопієві труби) відсутні, вагінальний
відросток – укорочений і сліпо закінчується. Похідні вольфових протоків (сім’явиносна
протока, сім’яні пухирці та придаток сім’яника) різною мірою гіпоплазовані. При
обох формах СТФ яєчка мають здатність достатньою мірою синтезувати тестостерон,
який на клітинному рівні метаболізується у дегідротестостерон. Однак дефект
андрогенових рецепторів у цитоплазмі призводить до відсутності або недостатності
відповіді андрогенів на стимул.
За умов повної форми захворювання відсутні усі ознаки, обумовлені дією
андрогенів. Зовнішні геніталії сформовані за жіночим типом. Яєчка можуть
знаходитись у черевній порожнині або пахових каналах, рідше – у великих статевих
губах, у гонадах виявляється гіперплазія клітин Лейдіга і Сертолі, сперматогенез
– відсутній. У пубертатному періоді формується жіночий фенотип, добре розвинені
молочні залози, однак статеве оволосіння відсутнє або дуже слабко виражене.
У дитячому віці показники росту відповідають нормативам для хлопчиків, кінцевий
зріст хворих перевищує нормативи для жінок.
Неповні форми СТФ клінічно гетерогенні. Будова зовнішніх статевих органів
відображає різний ступінь порушення дії андрогенів у внутрішньоутробний період.
У більшості дітей спостерігається мошонкова або пінеальна форма гіпоспадії,
характерна «шалеподібна» розщеплена мошонка, недорозвиток кавернозних тіл
статевого члена. Яєчка частіше розташовані у розщепленій мошонці або у пахових
каналах. У пубертатному періоді розвиваються молочні залози, оволосіння –
незначне, розвинене за жіночим типом, але у частини хворих симптоми
андрогенізації можуть бути активно виражені. Хворі – високорослі.
Варіант фенотипу, при якому будова зовнішніх статевих органів близька до
нормальних чоловічих, носить назву синдрому Рейфенштейна.
У всіх випадках синдрому тестикулярної фемінізації, за виключенням синдрому
Рейфенштейна, рекомендується вибір жіночої статі.
Хворі на СТФ мають високий ризик розвитку злоякісних пухлин яєчка.
Гормональні ознаки. Рівні ЛГ, тестостерону, дегідротестостерону підвищені.
Тест із введенням ХГ – позитивний. Тест із введенням тестостерону – негативний.
Рівень естрадіолу – дещо вищий, ніж у здорових хлопчиків. Зв’язування андрогенів
з фібробластами хворих in vitro знижене або відсутнє.
Дефіцит 5-α-редуктази (див. розділ «Порушення статевого диференціювання»)
Синдром дефіциту 5-α-редуктази є однією з форм несправжнього чоловічого
гермафродитизму. Хворі мають каріотип 46ХY; статевий хроматин – негативний.
Фермент 5-α-редуктаза каталізує перетворення тестостерону у дегідротестостерон,
який відіграє основну роль у процесах диференціювання зовнішніх статевих органів
у хлопчиків.
Дефіцит 5-α-редуктази виникає в результаті мутацій гену SRD5A2, локалізованому
на короткому плечі 2 хромосоми. Захворювання має аутосомно-рецесивний характер.
Клінічні ознаки. При народженні фенотип наближається до жіночого.
Урогенітальний синус вміщає короткий вагінальний відросток, який закінчується
сліпо. Статевий член нагадує гіпертрофований клітор, кавернозні тіла не
розвинені. Внутрішні статеві органи сформовані за чоловічим типом: яєчка
розвинені, розташовані у пахових каналах або скротолабіальних складках,
виявляються сім’явиносні протоки, сім’яні пухирці, придаток сім’яника.
Передміхурова залоза – недорозвинена.
У пубертатному віці підвищується концентрація тестостерону, що забезпечує
виражену маскулінізацію зовнішніх геніталій та розвиток вторинних статевих ознак
за чоловічим типом.
Гормональні ознаки. Рівні у крові тестостерону, ЛГ, ФСГ – нормальні,
рівень дегідротестостерону – низький. Співвідношення тестостерон/дегідротестостерон
суттєво підвищене (за рахунок зниження дегідротестостерону) і збільшується на
тлі стимуляційного тесту з ХГ (за рахунок підвищення рівня тестостерону).
Синдром резистентних яєчників
Характеризується яєчниковою недостатністю, яка пов’язана з нечутливістю
рецепторів яєчників до гонадотропінів. Причиною можуть бути генетичні дефекти
рецепторів ЛГ і ФСГ, а також аутоімунне ураження рецепторів.
Для хворих на синдром резистентних яєчників найтиповішим є нормальний жіночий
каріотип – 46ХХ. Лише у поодиноких випадках зустрічаються такі мозаїчні варіанти:
45ХО/47ХХХ, 46ХХ/47ХХХ.
Клінічні ознаки. Хворі мають нормальний або високий зріст, євнухоїдну
будову тіла, інфантильну зовнішність, незначне статеве оволосіння, нерозвинені
молочні залози. Характерні первинна або вторинна аменорея.
Зовнішні статеві органи сформовані за жіночим типом, недорозвинені. Матка та
яєчники суттєво зменшені у розмірах. При гістологічному дослідженні яєчників
спостерігається потовщення та фіброз білкової оболонки, велика кількість
примордіальних фолікулів у склерозованому корковому шарі, іноді – атретичні
фолікули. При кольпоцитологічному дослідженні виявляються ознаки гіпоестрогенії.
У частини хворих зберігається деяка функціональна активність яєчників. Такі
дівчата мають нормальний зріст і будову тіла. Статеве оволосіння у них достатньо
розвинене. Молочні залози самостійно розвиваються до 1-2 ступеня за Таннером.
Для таких хворих характерна вторинна аменорея. Матка та яєчники дещо зменшені у
розмірах. При гістологічному дослідженні спостерігається сполучнотканинне
переродження яєчників, у яких відмічаються примордіальні та кістозно змінені
фолікули, старі жовті, білі та фіброзні тіла.
Гормональні ознаки. У більшості випадків виявляється підвищення рівнів
ФСГ та ЛГ, зниження рівня естрадіолу у крові. У хворих з частково збереженою
функціональною активністю яєчників рівні ЛГ і ФСГ можуть бути нормальними, а
естрогени – незначно зниженими. Іноді спостерігається підвищення тільки ФСГ при
зниженні естрадіолу.
Діагноз підтверджується, якщо у біоптаті виявляються тільки примордіальні
фолікули, а зрілі фолікули – відсутні. Классификая и примеры формулировки диагноза
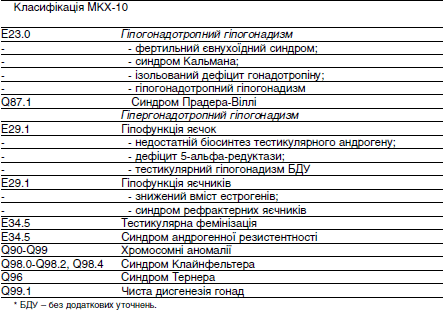
Класифікація МКХ-10
Приклади формулювання діагнозу
1) Гіпогонадотропний гіпогонадизм, синдром Кальмана, двосторонній паховий
крипторхізм.
2) Синдром тестикулярної фемінізації, повна форма, нечутливість до андрогенів.
3) Синдром Клайнфельтера, гіпергонадотропний гіпогонадизм. ДиагностикаКлінічні методи
При опитуванні з’ясовують родинний анамнез (особливості статевого
розвитку і росту батьків, найближчих родичів, перебіг вагітності та пологів у
матері, наявність або відсутність будь якої спадкової родинної хвороби,
соціальний статус родини, психологічний клімат у родині), швидкість росту та
збільшення ваги пацієнта, наявність перенесених або супутніх хронічних хвороб,
вживання лікарських засобів, перенесення хірургічних операцій, променевої або
хіміотерапії, наявність або відсутність скарг на нюх, слух, зір (наприклад:
гіпоосмія або аносмія при синдромі Кальмана, порушення слуху при синдромі
Шерешевського-Тернера, порушення зору при септооптичній дисплазії тощо).
При медичному огляді звертають увагу на розвиток і будову зовнішніх статевих
органів, наявність або відсутність вторинних статевих ознак, оцінюють їх
розвиток за Танером.
Проводять антропометричні вимірювання. Оцінюють зріст, вагу, звертають увагу на
співвідношення верхньої та нижньої частин тіла, довжину і будову кінцівок (наприклад:
порушення пропорцій тіла при синдромі Клайфельтера, вальгусні деформації
суглобів при синдромі Шерешевського-Тернера, Нунан та ін.).
Звертають увагу на наявність або відсутність характерних ознак вроджених і
спадкових захворювань – стигм дисембріогенезу (наприклад: характерний фенотип
при синдромах Шерешевського-Тернера, Нунан, тернероїдна форма змішаної
дизгенезії гонад).
Гормональні дослідження
- Визначення базальних рівнів ЛГ, ФСГ, пролактину в крові.
- Визначення базальних рівнів тестостерону, дегідроепіандростерону-сульфату,
дегідротестостерону, естрадіолу в крові (у хлопчиків).
- Визначення базальних рівнів естрадіолу, прогестерону, тестостерону,
дегідроепіандростерону-сульфату, дегідротестостерону в крові (у дівчат).
- Визначення добового рівня екскреції 17-КС з сечею.
- Проведення одного або декількох (при отриманні сумнівного результату одного з
тестів або для точнішої диференціальної діагностики) функціональних
стимуляційних тестів:
1) триденний тест з ХГ (у хлопчиків);
2) тест з гонадореліном (оцінка секреції ЛГ і ФСГ). Інформативний при досягненні
у підлітка кісткового віку 11 років (дівчата), 12 років (хлопчики);
3) тест з кломіфеном (оцінка секреції ЛГ і ФСГ). Інформативний при досягненні у
підлітка кісткового віку 11 років (дівчата), 12 років (хлопчики);
4) поєднаний тест з гонадореліном і кломіфеном;
5) тест з тестостероном (оцінка чутливості до андрогенів у хлопчиків);
6) тест з естрогенами і прогестероном (виключення або підтвердження первинної
аменореї, яка викликана гіпоплазією ендометрію);
7) тест з тироліберином (оцінка секреції пролактину).
- Оцінка імпульсної нічної секреції ЛГ.
- При підозрі на гіпопітуїтаризм – визначення базальних рівнів ТТГ, СТГ, АКТГ;
за необхідності – проведення функціональних стимуляційних тестів. Визначення
рівнів загального та вільного тироксину, кортизолу, ІРФ-1.
Інструментальні методи
- Встановлення кісткового віку за рентгенограмою кистей рук.
- УЗД яєчників, матки – оцінюють їх розміри та структуру, виявляють або
виключають аномалії їх розвитку.
- Генітографія дозволяє виявити матку та яєчники або їх відсутність; оцінити їх
розміри та форму, виявити аномалії розвитку внутрішніх статевих органів дівчат.
- УЗД наднирників, черевної порожнини, нирок дозволяє виявити або виключити
супутні вади розвитку сечостатевої системи.
- Селективна тестикулярна венографія дозволяє уточнити відсутність або наявність
яєчок та місце їх розташування.
- Рентгенограма черепа, КТ/МРТ головного мозку дозволяє виявити або виключити
пухлини та інші органічні ураження ЦНС (наприклад, дефекти серединних структур
головного мозку).
- Лапароскопія/лапаротомія з біопсією тканини гонад дозволяє уточнити наявність
або відсутність гонад, місце їх розташування, виключити або виявити дизгенезію
та злоякісні новоутворення гонад.
Генетичні методи
- Визначення статевого хроматину.
- Каріотипування.
Інші методи
- Кольпоцитологічне дослідження дозволяє оцінити естрогенну
насиченість організму у дівчат.
- Офтальмологічне дослідження (звуження полів зору, атрофія дисків зорових
нервів – додатковий показник ступеня порушення хіазми зорових нервів при
новоутвореннях; пігментний ретиніт – характерна ознака синдрому
Лоренса-Муна-Барде-Бідля; порушення кольорового зору, наявність колобом сітківки
– характерно для синдрому Кальмана, порушення зору, ністагм, мікрофтальмія та
гіпоплазія зорових нервів вказують на септооптичну дисплазію).
- Дослідження сперматограми у хлопчиків 16 років і старших.
- Проведення денситометрії у хворих пубертатного віку і старших (наявність
остеопорозу є додатковою ознакою гіпогонадизму).
Консультації спеціалістів
- Ендокринолог – визначення тактики лікування.
- Гінеколог – оцінка розвитку зовнішніх і внутрішніх статевих органів у дівчат,
виключення/підтвердження супутніх вад їх розвитку.
- Окуліст – виключення/підтвердження офтальмологічної патології як додаткового
критерію у диференціальній діагностиці.
- Генетик – виключення/підтвердження спадкової або вродженої патології, генних
та хромосомних аберацій тощо.
- Уролог – виключення/підтвердження супутніх вад розвитку сечостатевої системи.
- Психолог – виявлення відчуття неповноцінності, його глибини, патологічного
формування особистості у підлітків.
- За наявності патології інших органів та систем – консультації відповідних
спеціалістів (нейрохірург, хірург-ендокринолог, кардіолог, невропатолог,
отоларинголог, гастроентеролог, ортопед).
Диференціальна діагностика
Диференціальна діагностика проводиться, в основному, між окремими
формами гіпергонадотропного та гіпогонадотропного гіпогонадизму й між
гіпогонадизмом та конституціональною затримкою статевого дозрівання (див. розділ
«Затримка статевого дозрівання»).
Основною відмінністю між гіпергонадотропним та гіпогонадотропним гіпогонадизмом
є рівень гонадотропінів. Уже на початку пубертатного періоду при первинному
гіпогонадизмі рівні ЛГ та ФСГ суттєво підвищені. За умов важкого ураження гонад
у період ембріогенезу високі («посткастраційні») рівні гонадотропінів
виявляються у немовлят, що є важливим критерієм для виявлення синдрому анорхії у
хлопчиків (див. розділ «Крипторхізм»).
У хлопчиків широко застосовується тест з ХГ. За наявності первинного
гіпогонадизму тест з ХГ – негативний.
При багатьох синдромах, що мають яскраву симптоматику, діагноз гіпогонадизму
можна передбачити задовго до початку пубертатного періоду – синдром
Шерешевського-Тернера, Нунан, Прадера-Віллі, Лоуренса-Муна-Барде-Бідля, змішана
дизгенезія гонад, тяжкі форми порушення стероїдогенезу в наднирниках, множинний
дефіцит тропних гормонів гіпофізу тощо.
Для диференціальної діагностики між затримкою статевого дозрівання та
ізольованими формами гіпогонадотропного гіпогонадизму у пубертатному віці
важливе значення має стимулюючий тест з гонадореліном. За умов затримки
статевого дозрівання у відповідь на введення гонадореліну рівні ЛГ та ФСГ у
крові суттєво підвищуються (> 10 Од/л), за умов гіпогонадотропного гіпогонадизму
виявляється несуттєве підвищення гонадотропінів (< 10 Од/л), а при глибокому
тотальному дефіциті гонадотропінів рівні ЛГ та ФСГ не підвищуються взагалі.
У дівчат конституціональну затримку статевого дозрівання слід диференціювати з
затримкою статевого розвитку при недостатності 17-α-гідроксилази (одна з форм
вродженої дисфункції кори наднирників, яка не супроводжується вірилізацією). В
цьому випадку реєструються підвищені рівні прегненолону та прогестерону у крові;
рівні всіх 17-гідроксильованих стероїдів у сечі знижені, а їх секреція у
відповідь на введення ХГ або АКТГ відсутня чи ослаблена (порушення
17-гідроксилазної активності 17-α-гідроксилази). При порушенні 17,20-ліазної
активності 17-α-гідроксилази рівнь прегненолону нормальний або незначно
підвищений, рівні 17-гідроксипрогестерону, 17-гідроксипрегненолону та
прогестерону підвищені. У більшості хворих з недостатністю 17-α-гідроксилази має
місце артеріальна гіпертензія. Крім того, гіпогонадизм як у хлопчиків, так і у
дівчат розвивається і при інших формах вродженої дисфункції кори наднирників (диференційну
діагностику між окремими формами дивись у розділі «Вроджена дисфункція кори
наднирників»).
Ізольовану затримку менархе у дівчат диференціюють з повною формою синдрому
тестикулярної фемінізації. Для останнього характерні високорослість, відсутність
матки та яєчників, негативний статевий хроматин, каріотип 46ХY.
За наявності ізольованої затримки менархе слід виключити гіпоплазію ендометрію.
Відсутність крововиливу на тлі стимуляційної проби спочатку з прогестероном, а
потім – з естрогенами та прогестагенами вказує на гіпоплазію ендометрію. ЛечениеГормональна терапія гіпогонадизму носить замісний характер, її основною
метою є:
- Стимуляція розвитку вторинних статевих ознак.
- Стимуляція розвитку і росту зовнішніх та внутрішніх статевих органів.
- Формування будови тіла і фенотипу відповідно до статі.
- Забезпечення у постпубертатному періоді статевої функції, психосоціальної
адаптації, а за можливості – фертильності хворих.
- Активація процесів мінералізації кісткової тканини, профілактика остеопорозу.
У дівчат терапія гіпергонадотропного і гіпогонадотропного гіпогонадизму
проводиться однаково.
Лікування починають при досягненні у дівчини кісткового віку 11-12 років з
введення етинілестрадіолу у дозі 0,02-0,1 мг на добу впродовж 6 місяців, після
чого доза поступово може бути підвищена до 0,2-0,3 мг на добу.
Замість етинілестрадіолу можна використовувати препарат кон’югованих естрогенів
у дозі 625 мкг на добу, похідне естрадіолу валеріату 1 мг на добу, пластир з
естрадіолом по 0,05-0,1 мг 1-2 рази на тиждень, естрогенові гелі.
Монотерапію естрогенами краще починати з використання мінімально ефективних доз
із подальшим їх підвищенням.
Через 1-1,5 року монотерапії естрогенами починають замісне циклічне лікування
естрогенами та прогестагенами.
Етинілестрадіол приймають всередину по 0,05 мг на добу з 1 по 20 число кожного
місяця, з 21 по 26 число кожного місяця застосовують прегнін по 10 мг 3 рази на
день сублінгвально.
У дівчат 16 років і старших можна застосовувати наступну схему лікування:
протягом перших 16-20 днів кожного місяця застосовують естрогени (етинілестрадіол
по 0,05-0,1 мг 2 рази на день), у другій фазі циклу – гестагенні препарати
(прогестерон по 5-10 мг на день в/м або етістерон по 10-30 мг сублінгвально
тричі на день).
Застосовується також наступна схема лікування: естрогени приймаються кожного дня
з 1 по 21 число кожного місяця. З 12 або 15 числа по 21 число призначаються
прогестагени (медроксіпрогестерону ацетат по 5-10 мг на добу, всередину, або
норетілстерон 5 мг на добу всередину). З 22 числа і до кінця місяця приймання
всіх препаратів припиняється. З 1 числа наступного місяця цикл лікування
повторюють.
У наш час для замісної циклічної терапії з успіхом застосовуються комбіновані
естроген-гестагенні препарати.
У дівчат, хворих на синдром Шерешевського-Тернера, тернероїдну форму змішаної
дизгенезії гонад та множинний дефіцит тропних гормонів гіпофізу, лікування
гіпогонадизму починають у більш старшому віці, після проведення терапії
рекомбінантним людським гормоном росту – таким чином досягається максимальна
реалізація ростових можливостей хворої дитини.
У дівчат з синдромом Шерешевського-Тернера до початку естрогенотерапії необхідно
дослідити рівень гонадотропних гормонів, аби переконатися у відсутності
можливого спонтанного пубертату. Високі показники ЛГ і ФСГ дозволяють призначити
замісну терапію естрогенами.
У хлопчиків, хворих на гіпергонадотропний гіпогонадизм, лікування
призначається при досягненні кісткового віку 13-13,5 років. У деяких випадках,
коли гіпогонадизм супроводжується раннім формуванням євнухоїдних пропорцій тіла,
а також прогнозується виражена високорослість, лікування починають при
досягненні
12-річного кісткового віку.
Призначаються переважно пролонговані препарати естерів тестостерону, які
вводяться в/м 1 раз на місяць. Первинна доза препарату становить 50 мг у перший
рік лікування. На другий рік лікування доза збільшується до 100 мг; на третій
рік лікування доза становить 200-250 мг. У подальшому доза препарату коригується
згідно з віком пацієнта.
Окрім препаратів пролонгованої дії, можна використовувати тестостерону
андеканоат (жирнокислотний ефір природного тестостерону); підтримуюча доза
становить 80-120 мг на добу, приймається всередину у вигляді капсул,
розподіляється на 2 прийоми. Або призначається у вигляді ін’єкцій підліткам
16-18 років із задовільним прогнозом росту. Вводиться у дозі 1000 мг в/м один
раз на 10-12 тижнів.
У останні роки вважається, що препубертатний гіпогонадизм з вираженою
недостатністю статевого розвитку, особливо у випадках анорхії та значної
гіпоплазії яєчок, на перших етапах терапії потребує застосування ударних доз
андрогенів. У таких випадках використовується в/м по 250 мг 1 раз на місяць, або
тестостерон-енантат в/м по 150-200 мг 1 раз на 2-3 тижні. Лікування призначають
з 14-15 років хворим, які мають задовільний зріст і прогнозовану високорослість.
Терапія проводиться протягом 2-3 років. У подальшому доза препарату коригується
згідно з віком пацієнта. Враховуючи, що дія ефірів тестостерону з часом
знижується, пропонується чергувати ін’єкції андрогенів (упродовж 6 місяців) із
застосуванням нандролону по 50 мг на місяць протягом 3 місяців.
Юнакам, хворим на гіпогонадотропний гіпогонадизм, призначають препарати
ХГ (хорагон). Слід пам’ятати, що ХГ як монотерапію можна використовувати для
стимуляції статевого дозрівання тільки у підлітків з парціальними формами
гіпогонадизму. При важких формах, що супроводжуються тотальною відсутністю
гонадотропінів, терапія препаратами ХГ може виявитися неефективною або
потребувати дуже великих доз, які провокують резистентність до ХГ.
Препарати ХГ вводяться в/м 2 рази на тиждень упродовж 1,5-2 місяців з перервою
протягом місяця. Первинна доза становить 1000-1500 МО на ін’єкцію. За
недостатності ефекту через 6 місяців доза може бути підвищена до 2000-3000 МО.
Лікування триває 1-2 роки.
У перервах між курсами лікування ХГ рекомендуються препарати тестостерону у
малих дозах: тестостерон-енантат по 50-100 мг 1 раз на 2 тижні в/м, тетрастерон
– 125 мг 1 раз на місяць в/м, тестостерону андеканоат – 80 мг на добу всередину,
местеролон – 25-50 мг на добу всередину. До 14-річного віку у перервах між
курсами ХГ рекомендується призначати анаболічні стероїди у вікових дозах.
Після 17-18 років переходять на замісну андрогенотерапію препаратами
тестостерону у вікових дозах.
У дорослих пацієнтів для стимуляції сперматогенезу додатково призначають
препарати, які мають сполучену ЛГ та ФСГ активність (менотропін, урофолітропін
тощо).
У підлітків з гіпоталамічною формою гіпогонадизму (синдром Кальмана) ефективним
є імпульсне введення аналогів ЛГ-РГ (гонадорелін). Препарат вводиться за
допомогою спеціальної помпи-дозатора, що забезпечує періодичне введення
гонадореліну кожні 90 хвилин.
Умови, в яких треба надавати медичну допомогу
Поліклінічні відділення, в яких під наглядом ендокринолога проводять діагностику
і лікування гіпогонадизму, здійснюють довготривалий нагляд за хворими.
Комплексне обстеження хворого для підтвердження діагнозу, диференціальної
діагностики, вибір тактики лікування проводять під наглядом
педіатра-ендокринолога у ендокринологічних відділеннях, спеціалізованих центрах,
диспансерах, в яких здійснюють відповідний обсяг обстежень і лікувальних заходів.
Можливі результати надання медичної допомоги
У результаті лікування у підлітків з’являються вторинні статеві ознаки,
розвиваються зовнішні та внутрішні статеві органи.
Рекомендації щодо подальшого, у разі потреби, надання медичної допомоги
хворому.
Хворі на гіпогонадизм потребують ретельного довготривалого диспансерного
нагляду.
Нагляд ендокринолога відбувається 1 раз у 6 місяців, аж до завершення
пубертатного періоду. Один раз на рік – огляд окуліста, невропатолога,
психолога, гінеколога або андролога.
Рентгенограма кистей рук здійснюється 1 раз на рік. Рівень гонадотропних і
статевих гормонів вимірюється 1 раз на рік.
У разі розвитку гіпогонадизму хворим призначається відповідна гормональна
терапія.
Вимоги до дієтичних призначень та обмежень
Хворим надають рекомендації щодо дієти, збалансованої за вмістом білків,
жирів, вуглеводів, мікроелементів та вітамінів. При затримці росту і фізичного
розвитку призначається дієта, збагачена білками та вітамінами.
У разі поєднання ЗСД з ожирінням призначається низькокалорійна дієта з
обмеженням тугоплавких жирів та легкозасвоюваних вуглеводів. Юридические аспектыФорма інформованої згоди
Загальна добровільна форма інформованої згоди на медичне втручання, яку
погоджують підписом батьки або опікуни неповнолітніх дітей або повнолітні
пацієнти.
Методологія моніторингу та критерії ефективності виконання протоколу
Ендокринологи поліклінік, диспансерів, ендокринологічних центрів проводять
ретельний облік хворих на затримку статевого дозрівання, слідкують за
проведенням контрольних обстежень, забезпеченням пацієнтів лікарськими засобами.
Вартісні характеристики протоколу надають планові відділи спеціалізованих
центрів, диспансерів, лікарень.
|
|
{kind=link}